
Retinitis Pigmentosa
What is retinitis pigmentosa?
Retinitis Pigmentosa (RP) refers to a group of inherited diseases causing retinal degeneration. The cell-rich retina lines the back inside wall of the eye. It is responsible for capturing images from the visual field. People with RP experience a gradual decline in their visual because photoreceptor cells (rods and cones) die. Forms of RP and related disease include Usher syndrome, Leber’s congenital amaurosis, rode-cone disease, Bardet-Biedl syndrome, and Refsum disease, among others.
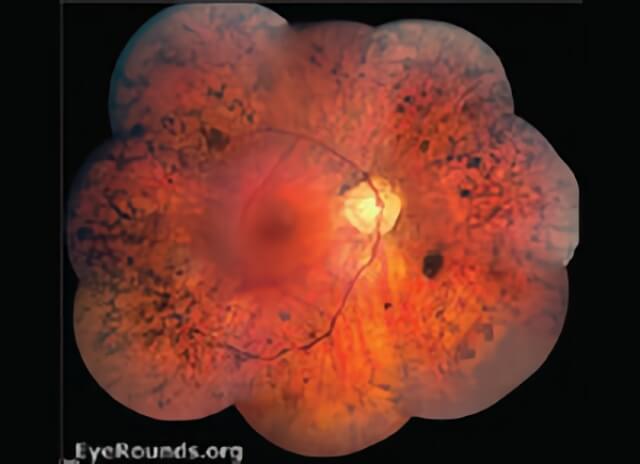
Retinitis Pigmentosa is shown in this photograph.
What are the symptoms of retinitis pigmentosa?
Symptoms depend on whether rods or cones are initially involved. In most forms of RP, rods are affected first. Because rods are concentrated in the outer portions of the retina and are triggered by dim light, their degeneration affects peripheral and night vision- become involved, the loss is in the color perception and central vision.
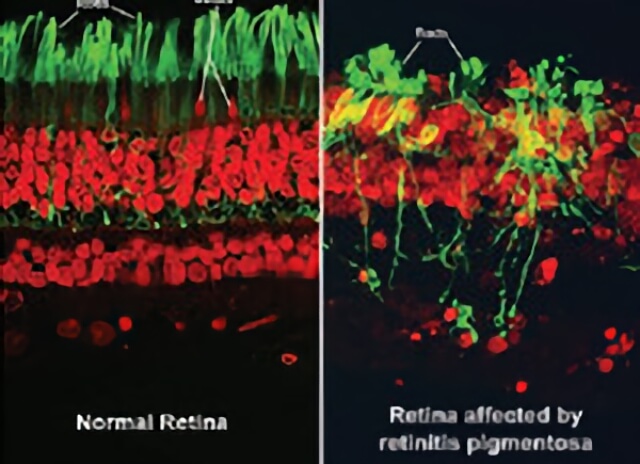
(Left side) shows the rods and cones of a normal healthy retina. (Right side) shows the rods and cones of a retina with retinitis pigmentosa.
How is retinitis pigmentosa inherited?
An estimated 100,000 people (1 in 4000) in the United States have RP, mainly caused by gene mutations (variations) inherited from one or both parents. Mutated genes give the wrong instructions to photoreceptor cells, telling them to make an incorrect protein, or too little or too much protein. (Cells need the proper amount of particular proteins in order to function properly.) Many different gene mutations exist in RP. In Usher syndrome, for example, at least 14 disease-causing genes have been identified. Genetic mutations can be passed from parent to offspring through one of three genetic inheritance pattern- autosomal recessive, autosomal dominant, or X-linked.
In autosomal recessive RP, both parents carry one copy of the mutated gene but have no symptoms themselves. Children have 25 percent chance of being affected by inheriting a mutated copy from each parent.
In autosomal dominant RP, usually one parent is affected and is the only parent with a mutated gene. A child has 50 percent chance of being affected by inheriting the mutated gene from the parent. In families with X-linked RP, the mother carries the mutated gene, and her sons have a 50 percent chance of being affected. Daughters are carriers and aren’t usually affected. However, some daughters are affected, but with milder symptoms.
Do all RP patients become blind?
No. The majority of patients retain good central vision throughout their life. Those who present with vision loss later in life will hold on to a substantial portion of their vision for the balance of their lives. Rarely patients can lose all vision, and approximately a quarter can become legally blind.

Normal vision without Retinitis Pigmentosa.

Vision as seen by a person with retinitis pigmentosa.
Are there any treatments?
RP patients can develop a form of retinal swelling known as cystoid macular edema that can be improved with fairly simple treatments. There is increasing evidence that anti-oxidants and a diet rich in anti-oxidants can help decrease the rate of vision loss. Vitamin A, Lutein, and DHA (fish oil) have been thought be to the most effective. More complex therapies are the goal of scientists and researchers studying gene therapy and microchip implants.
What testing is available for retinitis pigmentosa?
Electrophysiology: ERG is the most critical diagnostic test for RP because it provides an objective measure of rod and cone function across the retina and is sensitive to even mild photoreceptor impairment. The full-field ERG in RP typically shows a marked reduction of both rod and cone signals, although rod loss generally predominates. The ERG is usually abnormal by early childhood, except for some of the very mild and regional forms of RP. An ERG is usually performed periodically to track the progression of RP.
Electro-oculogram (EOG): Findings are always abnormal when ERG findings are abnormal; therefore, EOG is not helpful to the clinician in diagnosing RP. Central macular changes, normal ERG findings, and abnormal EOG findings are usually found amongst other macular dystrophy disorders.
Genetic: Genetic testing is available for RP. It helps assess the risk of passing the disorder from parent to offspring. It also helps with attaining an accurate diagnosis. A patient with an accurate diagnosis is in a better position to keep track of new findings, research developments, and treatment approaches. If a family member is diagnosed with RP, it is strongly advised that the other members of the family also have an eye exam by a physician who is specially trained to detect and treat retinal degenerative disorders.
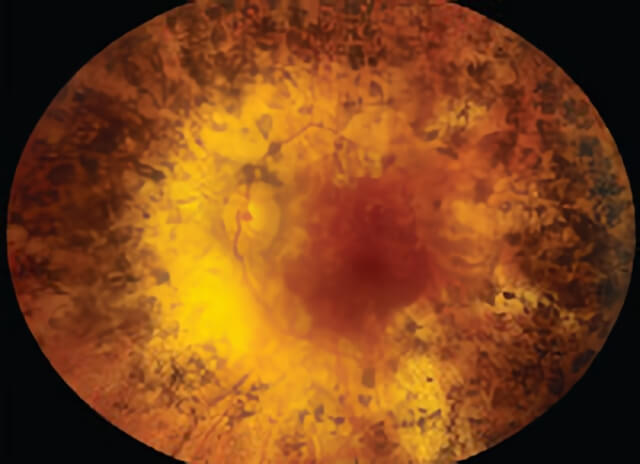
Color montage photo of a patient with retinitis pigmentosa.
Schedule Retinitis Pigmentosa Treatment in Northern California with Retinal Consultants Medical Group
Since 1977, Retinal Consultants Medical Group has been providing outstanding care to patients throughout Northern California, including Sacramento, Modesto, and Stockton. Our retina specialists and surgeons treat multiple vitreoretinal conditions, such as age-related macular degeneration and diabetic retinopathy. We invite you to contact us with any questions or schedule an appointment today.