
Congenital X-Linked Retinoschisis
What is congenital X-linked retinoschisis?
Congenital X-linked retinoschisis is an early-onset hereditary retinal disease characterized by splitting (schisis) of the retinal layers, particularly in the center of vision (fovea) and/or in the peripheral retina.
Estimates indicate that 1 in 5000 to 1 in 25,000 patients experience congenital X-linked retinoschisis depending on which population is studied.
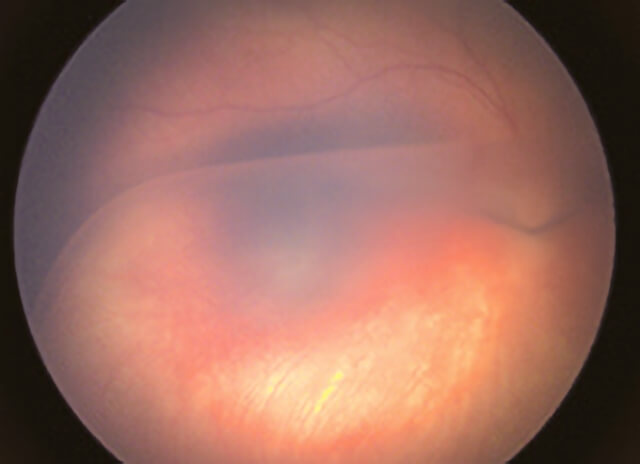
The peripheral retina also may be involved, with splitting of the retinal layers resulting in a blister-like elevation of the lower part of the retina.
Learn more about congenital X-linked retinoschisis at ASRS
What are the symptoms of congenital X-linked retinoschisis?
Vision loss is the most common symptom. Congenital X-linked retinoschisis is typically diagnosed in males between the ages of 3 months and school age with symptoms that include crossed eyes (strabismus), abnormal eye movements (nystagmus) and “lazy eye” (amblyopia). When loss of vision is mild, it is often not detected until a child fails vision screening at school.
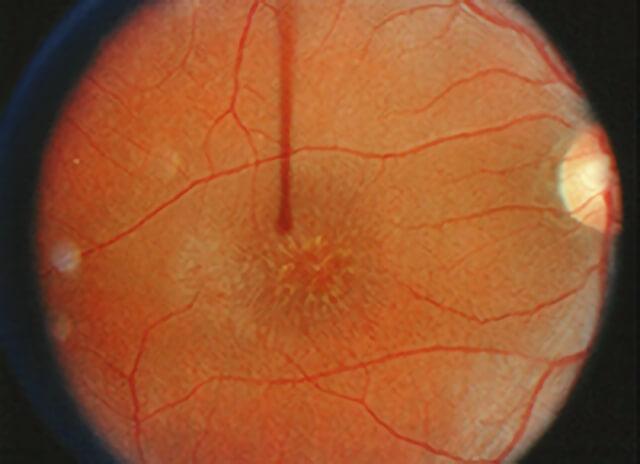
Congenital x-linked retinoschisis remains a clinical diagnosis. Affected males typically have characteristic foveal schisis.
What are the causes of congenital X-linked retinoschisis?
The exact cause of the retinal-splitting characteristic of congenital X-linked retinoschisis has yet to be established. Identification of at least 191 different mutations in the retinoschisin (RS1) gene—an adhesive protein—suggests that the condition occurs as a consequence of impaired cell-to-cell adhesion or bonding. Apart from heredity, there are no specific risk factors.
How is congenital X-linked retinoschisis diagnosed and tested?
Congenital X-linked retinoschisis remains a clinical diagnosis. Affected males typically have characteristic foveal schisis. The peripheral retina also may be involved, with splitting of the retinal layers resulting in a blister-like elevation of the lower part of the retina. In severe cases retinal detachment may occur. Congenital X-linked retinoschisis typically affects both eyes, although often not to the same degree.
An electroretinogram (ERG) test, which evaluates the electrical “circuitry” properties of the retina, shows a typical finding in patients with congenital X-linked retinoschisis (selective reduction of the amplitude of the b-wave). Optical coherence topography (OCT)—a way of imaging the retinal structure— demonstrates the typical intraretinal cystic spaces.
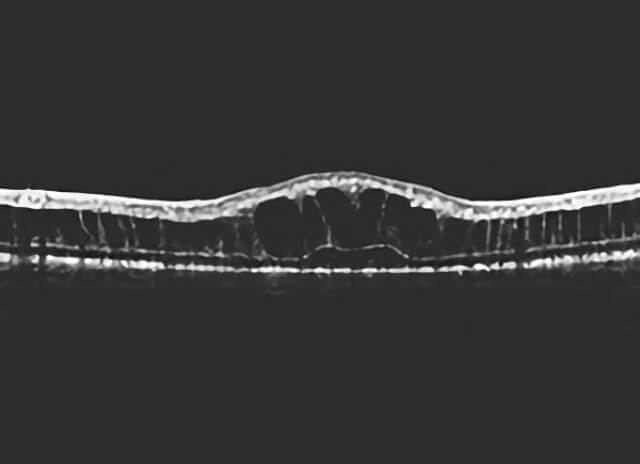
An electroretinogram (ERG) test, which evaluates the electrical “circuitry” properties of the retina, shows a typical finding in patients with congenital x-linked retinoschisis (selective reduction of the amplitude of the b-wave).
The trait is passed genetically to affected males by their mothers. Genetic testing is available and may prove helpful for women at risk of carrying the trait for RS. Female carriers usually cannot be identified clinically because they show no retinal findings and have normal ERG responses and OCTs. They are identified only through analysis of their family trees. Examination of male siblings and other maternal male relatives (maternal grandfather, maternal uncles, etc.) may be helpful. Genetic counseling is advisable for families in which congenital X-linked retinoschisis has affected male members.
How is congenital X-linked retinoschisis treated?
The foveal and peripheral retinal splitting that occurs in schisis cannot be treated, but secondary complications may require treatment. These include vitreous hemorrhage or bleeding into the gel that fills the back of the eye (4% – 40%) and retinal detachment (5%–22%). Blood filling the eye may impair vision to the degree that it needs to be removed to restore vision. The retinal detachments in congenital X-linked retinoschisis can be complicated, and are treated surgically as well. Crossed eyes (strabismus) and lazy eye (amblyopia) are treated by pediatric ophthalmologists.
Congenital X-linked retinoschisis is a lifelong disease. Vision is typically between 20/60 and 20/120 during teenage and middle-aged years. This level of vision can be improved with low-vision aids. Once stable, individuals with Congenital X-Linked retinoschisis are usually followed every 6-12 months with examination and imaging (OCT and color photography).
Information and images for Congenital X-Linked Retinoschisis is from the American Society of Retina Specialists
Copyright 2016 The Foundation of the American Society of Retina Specialists. All rights reserved.
Schedule Congenital X-Linked Retinoschisis Treatment in Northern California with Retinal Consultants Medical Group
Since 1977, Retinal Consultants Medical Group has been providing outstanding care to patients throughout Northern California, including Fairfield, Tracy, Sacramento, Modesto, and Stockton. Our retina specialists and surgeons treat multiple vitreoretinal conditions, such as age-related macular degeneration and diabetic retinopathy. We invite you to contact us with any questions or schedule an appointment today.